![]()
Symphonypy usage with your own reference without batch
[ ]:
!pip install scanpy[leiden] openTSNE symphonypy
!gdown "1jW548g6ERFS0t7NywgyjRs6VaE5QwXbg&confirm=t"
[2]:
import numpy as np
import seaborn as sns
import scanpy as sc
import symphonypy as sp
import matplotlib.pyplot as plt
sc.set_figure_params(dpi=80)
sns.set_style("ticks")
adata_ref = sc.read_h5ad("PBMC_Satija.h5ad")
adata_ref = adata_ref[adata_ref.obs.donor == "P1"]
adata_ref.obs.head()
[2]:
| orig.ident | lane | donor | time | celltype.l1 | celltype.l2 | celltype.l3 | Phase | |
|---|---|---|---|---|---|---|---|---|
| L1_AAACCCATCTGCGGAC | P1_0 | L1 | P1 | 0 | CD4 T | CD4 TCM | CD4 TCM_1 | S |
| L1_AAACGAAAGTTACTCG | P1_0 | L1 | P1 | 0 | CD4 T | CD4 TCM | CD4 TCM_3 | G1 |
| L1_AAACGCTAGGTCGTCC | P1_0 | L1 | P1 | 0 | CD8 T | CD8 TEM | CD8 TEM_2 | S |
| L1_AAAGAACGTATCCTCC | P1_0 | L1 | P1 | 0 | CD8 T | CD8 Naive | CD8 Naive | S |
| L1_AAAGAACTCGCTTAAG | P1_0 | L1 | P1 | 0 | CD4 T | CD4 TCM | CD4 TCM_1 | S |
Reference preprocessing and preparation for label transfer using Harmony. We recommend using the same normalization factor for reference and query datasets.
[3]:
sc.pp.normalize_total(adata_ref, target_sum=1e5) # 1e5 will be used for query
sc.pp.log1p(adata_ref)
sc.pp.highly_variable_genes(
adata_ref,
n_top_genes=3000,
)
adata_ref.raw = adata_ref
/usr/local/lib/python3.10/dist-packages/scanpy/preprocessing/_normalization.py:206: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
[4]:
adata_ref = adata_ref[:, adata_ref.var.highly_variable]
sc.pp.scale(adata_ref, max_value=10)
sc.pp.pca(adata_ref, n_comps=30, zero_center=False)
/usr/local/lib/python3.10/dist-packages/scanpy/preprocessing/_scale.py:299: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
Now let’s compute UMAP for futher visualization and its coordinates transfer, and perform clustering with Leiden algorithm.
[5]:
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
[6]:
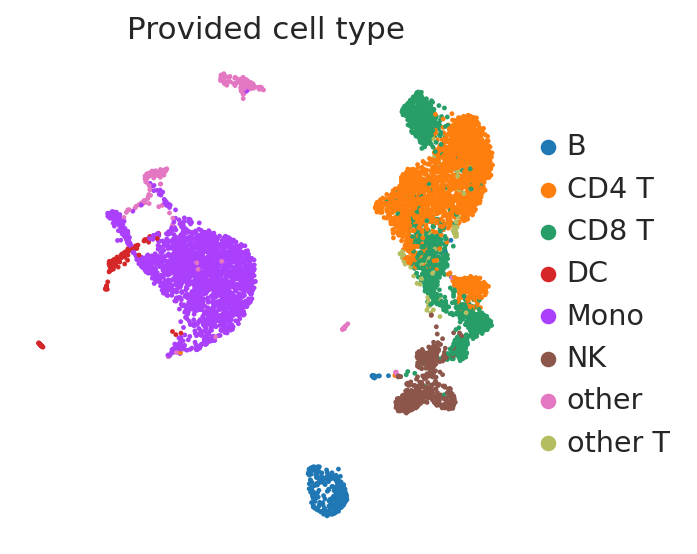
sc.pl.umap(
adata_ref,
color=["celltype.l1"],
frameon=False,
title=["Provided cell type"],
)
Query mapping and label transfer
[7]:
adata_query = sc.datasets.pbmc3k()
sc.pp.normalize_total(adata_query, target_sum=1e5)
sc.pp.log1p(adata_query)
Let’s do embedding mapping and label transfering with symphonypy.
[8]:
# Running Symphony
sp.tl.map_embedding(
adata_query=adata_query,
adata_ref=adata_ref
)
/usr/local/lib/python3.10/dist-packages/symphonypy/tools.py:307: UserWarning: Not found `harmony` object in adata_ref.uns.
Assuming that adata_ref doesn't have any batches, and using 'X_pca' representation of adata_ref for clustering.
Otherwise, firstly run symphonypy.pp.harmony_integrate on adata_ref to account for them.
warnings.warn(
WARNING:symphonypy:541 out of 3000 genes from the reference are missing in the query dataset or have zero std in the reference, their expressions in the query will be set to zero
[9]:
# Mapping UMAP coordinates
sp.tl.ingest(
adata_query=adata_query,
adata_ref=adata_ref,
use_rep="X_pca_harmony",
)
/usr/local/lib/python3.10/dist-packages/umap/umap_.py:1945: UserWarning: n_jobs value 1 overridden to 1 by setting random_state. Use no seed for parallelism.
warn(f"n_jobs value {self.n_jobs} overridden to 1 by setting random_state. Use no seed for parallelism.")
/usr/local/lib/python3.10/dist-packages/symphonypy/_utils.py:372: UserWarning: 'X_pca' adata_reference's representation was used for reference, while 'X_pca_harmony' was used for query. Be sure that they correspond.
warnings.warn(
[10]:
# Labels prediction
sp.tl.transfer_labels_kNN(
adata_query=adata_query,
adata_ref=adata_ref,
ref_labels=["celltype.l1"],
ref_basis="X_pca",
query_basis="X_pca_harmony",
)
/usr/local/lib/python3.10/dist-packages/sklearn/neighbors/_classification.py:215: DataConversionWarning: A column-vector y was passed when a 1d array was expected. Please change the shape of y to (n_samples,), for example using ravel().
return self._fit(X, y)
[11]:
import matplotlib.pyplot as plt
fig, axes = plt.subplots(figsize=(8, 4), ncols=2)
sc.pl.umap(
adata_query,
color="celltype.l1",
frameon=False,
title="Query dataset",
ax=axes[0],
show=False,
legend_loc=None,
)
sc.pl.umap(
adata_ref,
color="celltype.l1",
frameon=False,
title="Reference dataset",
ax=axes[1],
show=False,
)
[11]:
<Axes: title={'center': 'Reference dataset'}, xlabel='UMAP1', ylabel='UMAP2'>

[12]:
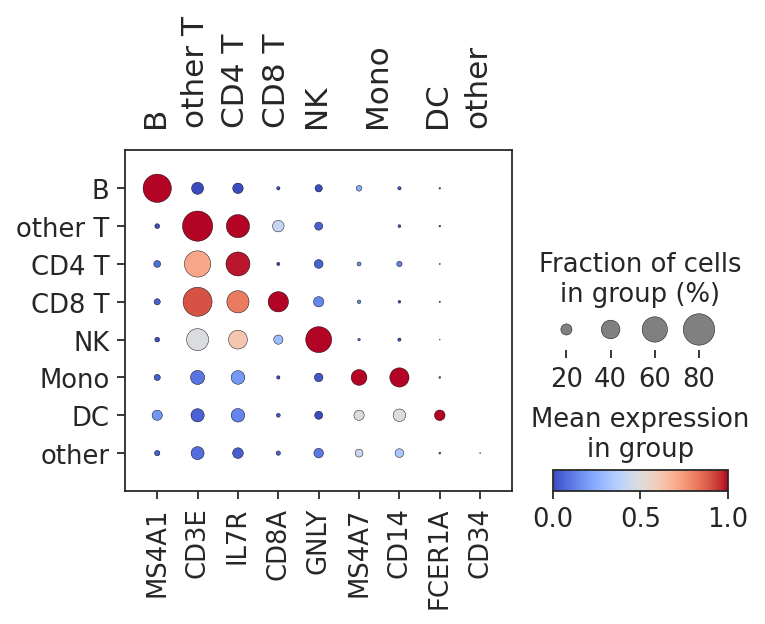
marker_genes = {
"B": ["MS4A1"],
"other T": ["CD3E"],
"CD4 T": ["IL7R"],
"CD8 T": ["CD8A"],
"NK": ["GNLY"],
"Mono": ["MS4A7", "CD14"],
"DC": ["FCER1A"],
"other": ["CD34"],
}
adata_query.obs["celltype.l1"] = (
adata_query.
obs["celltype.l1"].
cat.
reorder_categories(
list(marker_genes.keys())
)
)
sc.pl.dotplot(
adata_query,
var_names=marker_genes,
groupby="celltype.l1",
standard_scale="var",
dot_max=0.8,
cmap="coolwarm",
)